lisozimas:
Es una proteina de función enzimática que provoca efecto bacteriostático , o sea mantiene "a raya" la reproducción bacteriana, por eso está en nuestra saliva.
Y en condiciones de laboratorio ha demostrado inactivar el HIV, por lo cual la saliva no contagia el virus en cuestión.

La tripsina es secretada en el intestino delgado, donde actúa hidrolizando péptidos en sus componentes estructurales básicos conocidos como aminoácidos (éstos péptidos a su vez son el resultado de la actividad de la enzima pepsina, que degrada proteínas en el estómago). Esto es necesario para el proceso de absorción de las proteínas presentes en la comida, ya que a pesar de que los péptidos son mucho más pequeños con respecto a las proteínas, son aún demasiado grandes para ser absorbidos por la membrana del intestino . La tripsina realiza la hidrólisis de los enlaces peptídicos. El mecanismo enzimático es igual al de las otras Serín proteasas: una tríada catalítica convierte a la serina del sitio activo en nucleofílica. Esto se logra modificando el ambiente electrostático de la serina. La reacción enzimática catalizada por las tripsinas es termodinámicamente favorable pero tiene una alta energía de activación (es cinéticamente desfavorable). Las tripsinas tienen un pH óptimo de operación de 8 y una temperatura óptima de operación de 37 °C.
El residuo de aspartato (Asp 189) localizado en la región catalítica (S1) de las tripsinas tiene la función de atraer y estabilizar lisinas y argininas (ambas cargadas positivamente) y por ello es responsable de la especificidad de la enzima. Esto significa que las tripsinas predominantemente cortan proteínas en el extremo carboxílico (ó extremo C-terminal) de sus residuos de lisinas y argininas, excepto cuando el residuo siguiente es una prolina. Las tripsinas son endopeptidasas, esto es, el corte se lleva a cabo en medio de la cadena peptídica y no en los residuos terminales de la misma.
Principales enzimas digestivas
Enzima | Origen | Substrato | Función Catalítica o Productos |
Amilasa salival | Glándulas salivales | Almidón | Hidroliza LOS enlaces 1,4, produciendo dextrinas limitantes, matotriosa y maltosa. |
Pepsinas | Estómago | Proteínas y polipéptidos | Rompen los enlaces peptídicos adyacentes a los aminoácidos aromáticos |
Tripsina | Páncreas exocrino | Proteína y polipéptidos | Rompen los enlaces peptídicos adyacentes a la arginina o lisina. |
Quimotripsinas | Páncreas exocrino | Proteína y polipéptidos | Rompen los enlaces peptídicos adyacentes a los aminoácidos aromáticos |
Carboxipeptidasa | Páncreas exocrino | Proteína y polipéptidos | Separa los carboxiaminoácidos terminales |
Lipasa pancreática | Páncreas exocrino | Triglicéridos | Monogliceridos y ácidos grasos |
Esterasa pancreática | Páncreas exocrino | Esteres de colesterol | Colesterol |
Amilasa pancreática | Páncreas exocrino | Almidón | Igual que la amilasa salival |
Ribonucleasa | Pancreas exocrino | ARN | Nucleótidos |
Desoxirribonucleasa | Páncreas exocrino | ADN | Nucleótidos |
Enteropeptidasa | Mucosa intestinal | Tripsinógeno | Tripsina |
Aminopeptidasas | Mucosa intestinal | Polipéptidos | Separa el aminoácido N-terminal del peptido |
Dipeptidasas | Mucosa intestinal | Dipeptidos | Dos aminoácidos |
Maltasa | Mucosa intestinal | Maltosa, maltotriosa | Glucosa |
Lactasa | Mucosa intestinal | Lactosa | Galactosa y glucosa |
Sacarasa | Mucosa intestinal | Sacarosa | Fructosa y glucosa |
| Dextrinasa limitante | Mucosa intestinal | Dextrinas limitantes | Glucosa |
Nucleasa y enzimas relacionadas | Mucosa intestinal | Ácidos nucleicos (ADN y ARN) | Pentosas y bases púricas y pirimídicas |
Diversas peptidasas | Citoplasma de las células de la mucosa | Di-, tri-, y tetrapéptidos | Aminoácidos. |
Algunas hormonas gastrointestinales
Hormona | Tejido de origen | Tejido diana | Acción principal |
Gastrina | Estómago y duodeno | Células secretorias y músculos del estómago | Producción y secreción de HCL; estimulación de la movilidad gástrica (peristaltismo) |
| Colecistokinina-pancreozimina (CCK-PZ) | Intestino delgado anterior (duodeno) | Vesícula biliar | Contracción de la vesícula biliar |
Secreción jugo pancreático | |||
Secretina | Duodeno | Páncreas; células secretoras y músculos del estómago | Secreción de agua y NaHCO3 ; inhibición de la motilidad gástrica. |
Péptido inhibidor gástrico (GIP) | Intestino delgado anterior | Mucosa y musculatura gástrica | Inhibición de la secreción y motilidad gástrica, estimulación de las glándulas de Brunner |
| Bulbogastrona o enterogastrona | Intestino delgado anterior | Estómago | Inhibición de la secreción y motilidad gástrica |
Péptido vasoactivo intestinal (VIP) | Duodeno | Aumento del flujo sanguíneo; secreción de líquido pancreático acuoso; inhibición de secreción gástrica. | |
Enteroglucagon | Duodeno | Yeyuno, páncreas | Inhibición de la motilidad y secreción |
Encefalina | Intestino delgado | Estómago, páncreas, intestino | Estimulación de la secreción de HCL, inhibición de secreción de enzima pancreática y de la motilidad instetital |
Somatostatina | Intestino delgado | Estómago, páncreas, intestinos, arteriolas esplácnicas | Inhibición de la secreción de HCL, secreción pancreática, motilidad intestinal y flujo sanguíneo espláctico. |



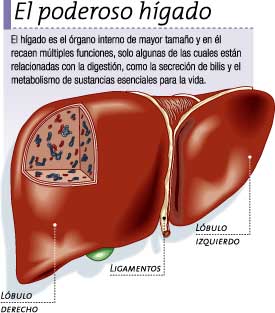









 El ser humano tiene 20 dientes que utiliza durante la fase inicial del desarrollo de los maxilares y que reciben el nombre de dientes de leche o de la infancia. A medida que los maxilares crecen, estos dientes son reemplazados por otros
El ser humano tiene 20 dientes que utiliza durante la fase inicial del desarrollo de los maxilares y que reciben el nombre de dientes de leche o de la infancia. A medida que los maxilares crecen, estos dientes son reemplazados por otros  Detrás de ésta existen dos dientes denominados premolares, con dos cúspides cada uno. Detrás de los premolares están el primero, el segundo y el tercer molar, que tienen una superficie de masticación relativamente plana, lo que permite triturar y moler los alimentos. Por lo general, la comida se corta con los dientes incisivos frontales, su tamaño se reduce por los
Detrás de ésta existen dos dientes denominados premolares, con dos cúspides cada uno. Detrás de los premolares están el primero, el segundo y el tercer molar, que tienen una superficie de masticación relativamente plana, lo que permite triturar y moler los alimentos. Por lo general, la comida se corta con los dientes incisivos frontales, su tamaño se reduce por los  En otros, el diente permanente puede estar impactado en el hueso, por lo que su erupción es imposible. También pueden existir dientes supernumerarios o adicionales. El alineamiento defectuoso o maloclusión se puede producir también después de la erupción. Debido a que la posición de un diente en la mandíbula no es estática, la pérdida de una pieza dentaria puede hacer que los dientes adyacentes se inclinen hacia el espacio vacío y el diente correspondiente del maxilar opuesto continúe su crecimiento en dicho espacio.
En otros, el diente permanente puede estar impactado en el hueso, por lo que su erupción es imposible. También pueden existir dientes supernumerarios o adicionales. El alineamiento defectuoso o maloclusión se puede producir también después de la erupción. Debido a que la posición de un diente en la mandíbula no es estática, la pérdida de una pieza dentaria puede hacer que los dientes adyacentes se inclinen hacia el espacio vacío y el diente correspondiente del maxilar opuesto continúe su crecimiento en dicho espacio. produce la infección del tejido de la cavidad pulpar que al final conduce a necrosis o formación de abscesos, que si no se detiene pueden llegar a afectar al maxilar. El proceso de las caries se acompaña de la formación de gases putrefactos. Si se obstruye la entrada en la cavidad pulpar, se produce un dolor severo a medida que aumenta la presión de los gases. En muchos casos, el diente se puede tratar con terapia del conducto radicular que elimina el material infectado que se encuentre en él. En los casos graves el diente se extrae.
produce la infección del tejido de la cavidad pulpar que al final conduce a necrosis o formación de abscesos, que si no se detiene pueden llegar a afectar al maxilar. El proceso de las caries se acompaña de la formación de gases putrefactos. Si se obstruye la entrada en la cavidad pulpar, se produce un dolor severo a medida que aumenta la presión de los gases. En muchos casos, el diente se puede tratar con terapia del conducto radicular que elimina el material infectado que se encuentre en él. En los casos graves el diente se extrae.














 son gemelos
son gemelos












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}